第254节 出生缺陷
<>,,。 <>%~%。。.%,。,。 <>。,,,。,,。 <>【】 <>,,、、、、、。 <>,。,,,,。,。 <><> <>,、。,;,。,,,,。,,。 <>,。,,。,。 <>。,。 <><> <>,,。,。。,。,。 <><> <>,,。。,,。,;,。 <><> <>,,,。,,。()。。
先天性心脏病
<>,。、、。,。()。()、。,,。 <>。,,。、、。,、。 <><>、 <>。 <>
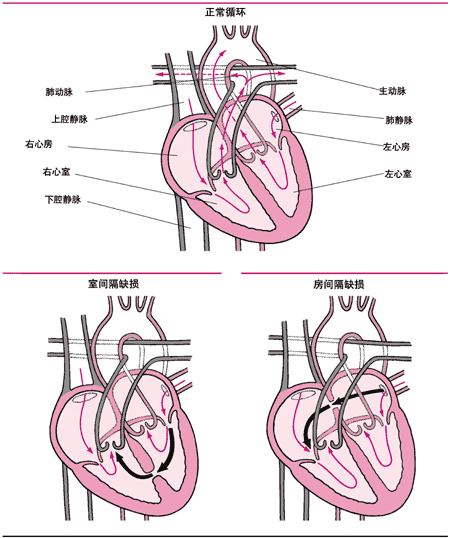 <>(),。(),。
<>、,。,,,,、、、。。,,。
<>、,。、。
<><>
<>()()。
<>,,,,。,。,~。,,,,、。
<>
<>(),。(),。
<>、,。,,,,、、、。。,,。
<>、,。、。
<><>
<>()()。
<>,,,,。,。,~。,,,,、。
<>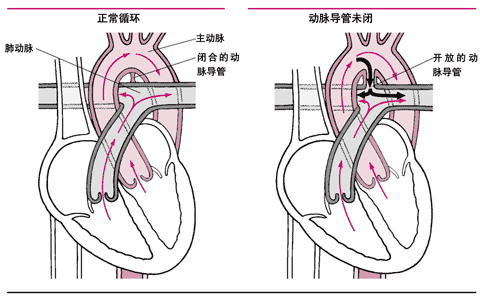 <>。,。,。,。
<>,,。,,。
<><>
<>,,()。
<>(),。,,。。,。。,,。,,,。()。,,。
<><>
<>,。
<>,,。
<>,。,,。,、、、。()。,。
<>,,、,,、,。,——。,,,,。。。
<><>
<>,、。
<>,。,,。,。,,。,,。
<>,,、、。,,。(~)。
<>,,,。()。,,。,,。。。、、、。
<><>
<>,,。
<>,,。,,,。,。
<>。,(),,,。
<>,、、、。。,。,,。,,,。
<><>
<>。。,。,,,,。。
<><>
<>
<>。,。,。,。
<>,,。,,。
<><>
<>,,()。
<>(),。,,。。,。。,,。,,,。()。,,。
<><>
<>,。
<>,,。
<>,。,,。,、、、。()。,。
<>,,、,,、,。,——。,,,,。。。
<><>
<>,、。
<>,。,,。,。,,。,,。
<>,,、、。,,。(~)。
<>,,,。()。,,。,,。。。、、、。
<><>
<>,,。
<>,,。,,,。,。
<>。,(),,,。
<>,、、、。。,。,,。,,,。
<><>
<>。。,。,,,,。。
<><>
<>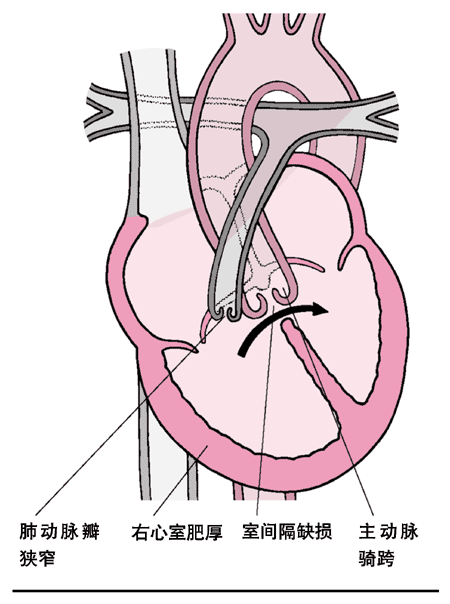 <>、()、()、。
<>。。,,,。,,;,;,。,。,,。,,,。
<>、()、()、。
<>。。,,,。,,;,;,。,。,,。,,,。
消化道畸形
<>:、、、、。,。。 <>
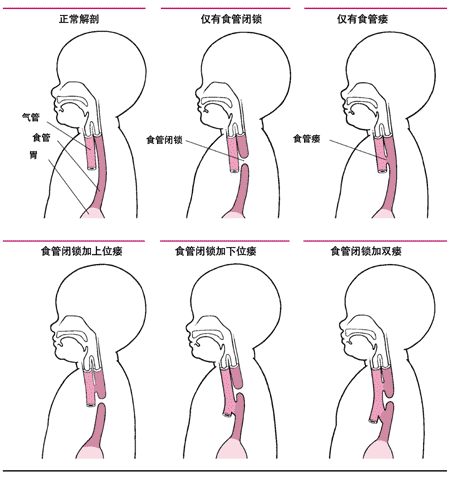 <>
<>,。。
<><>
<>。
<>,,。,。
<>,,()。,,。
<>,,。,,,。。
<><>
<>,。
<>,。、、。,。
<>,,,,。。
<>,,。
<><>
<>()。
<>。,,,。
<>,。。,()。,,。,。,,。
<><>
<>。
<>,。,。
<><>
<>。
<>,。,,,。
<>、、。。,。
<><>
<>。
<>,。,。,,,,,。
<>【】
<>,,,。~,、,(、、)。
<>。,,,。,,()。
<>【】
<>。,。%~%,,。,。,,。
<>
<>,。。
<><>
<>。
<>,,。,。
<>,,()。,,。
<>,,。,,,。。
<><>
<>,。
<>,。、、。,。
<>,,,,。。
<>,,。
<><>
<>()。
<>。,,,。
<>,。。,()。,,。,。,,。
<><>
<>。
<>,。,。
<><>
<>。
<>,。,,,。
<>、、。。,。
<><>
<>。
<>,。,。,,,,,。
<>【】
<>,,,。~,、,(、、)。
<>。,,,。,,()。
<>【】
<>。,。%~%,,。,。,,。
骨骼肌肉系统畸形
<>,、、、。。 <><> <>。,。()。 <>
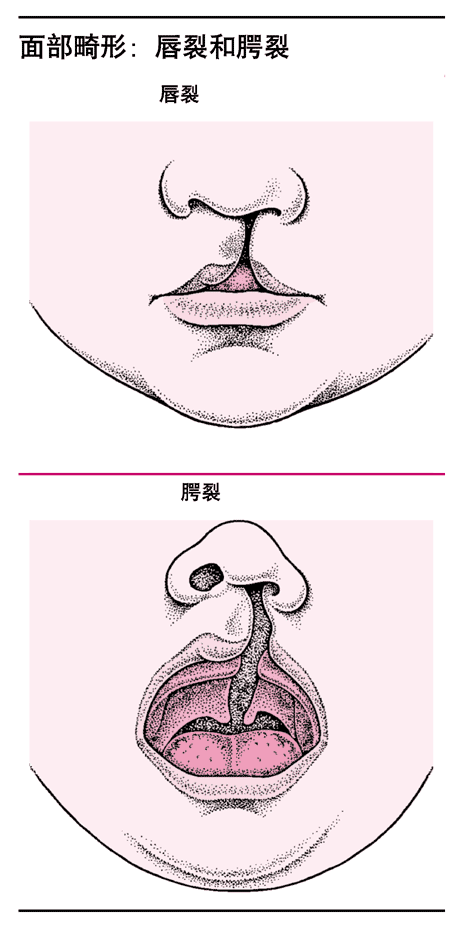 <>,,。。,/~/,/,/。
<>,。
<>。,,。。
<><>
<>,。,(),。
<>。,。,。,,。
<><>、
<>,。。、。。,,。
<>(—)(—)。,。
<>。,。,。
<>,,,。。,,。,。,,。
<>
<>,,。。,/~/,/,/。
<>,。
<>。,,。。
<><>
<>,。,(),。
<>。,。,。,,。
<><>、
<>,。。、。。,,。
<>(—)(—)。,。
<>。,。,。
<>,,,。。,,。,。,,。
<>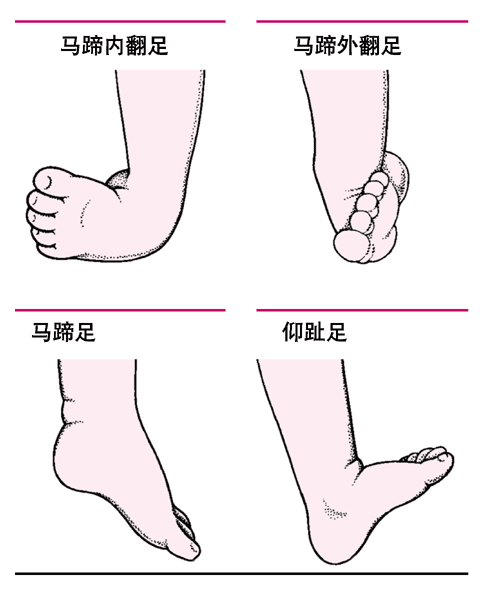 <>
<><>
<>()。
<>。,,,,。
<>“”,,。。。
<><>
<>。
<>,,,,。,。,。
<><>
<>,。
<>。、。,。
<><>
<>,,。
<>(- ),,,。。。
<>
<><>
<>()。
<>。,,,,。
<>“”,,。。。
<><>
<>。
<>,,,,。,。,。
<><>
<>,。
<>。、。,。
<><>
<>,,。
<>(- ),,,。。。
脑和脊髓畸形
<>,()。 <><> <> 。,。 <> 。,,。 <> 。,。 <> 。,。 <> 。,,。 <> ()。,,。,。。。。 <>。,(),。,。,。,,。,,,;。 <>-(-) <>,。 <><> <>。 <>,。/。 <>,。;()。 <>,。,。,。 <>、,。,、、。。 <>。,,,、。,,。,,。 <>
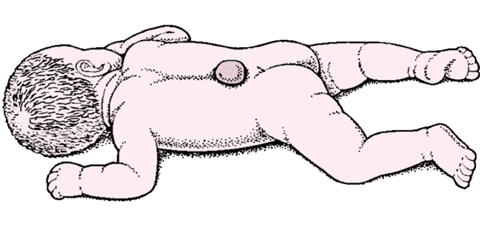 <>
<>
眼畸形
<> ,,。,、。 <> ()。、。,()。
肾脏和泌尿道畸形
<>。,。。 <><> <>,、、()、()。();,,()。 <>。,,,。(),,()。 <><> <>。,()。,。。,,,,。。 <><> <>/,,;,。,。。 <><> <>。 <>()。,,。,,,。 <>(),,。,(),;,。,。 <>(),。()。 <>,。,。,。
染色体异常
<>。,()。 <>: <>·; <>·; <>·。 <><> <>()。 <>,。,。%,%。。()。%,%~%。,/~/。 <>【】 <>。、,。(),,。 <>,,,,,,,,。,,。。%。 <>【】 <>,。,,。 <>,,。 <>【】 <>,,,,。,。(),。,。。,,,。,。 <><> <>。()。,。,,。,,,。,,,,。,()。,,。,。 <>,,,。,。。,,。 <><> <>()。 <>/。(),。 <>,(),,,,,。()()。,、、。。(),。 <>(),。,,,。 <><> <>,/。,,、。 <>。,。 <><> <>( )(),,/。 <>,,。,。,,。,,,,。,,。 <><> <>。,。,。 <><> <>,(),。。,。。 <>,、,,(),,,,,。,。
上一篇:第253节 新生儿和婴儿感染
下一篇:第255节 精神发育迟滞