第十一节 心肌病
近年来,临床和病理学家将原因不明而又非继发于全身或其他器官系统疾病的心肌原发性损害定名为原发性心肌病(primary cardiomyopathy)。它是非风湿性、非高血压性、非冠状动脉性心肌结构和功能的病理改变。其病理过程属于代谢性而非炎症性,在发病机制上与其它已知病因引起的心脏病无关。相反,若心肌病变与已知病因有关,或继发或伴发于某种全身性疾病时,则称为继发性心肌病(secondary cardiomyopathy)。原发性心肌病较少见,但分布于世界各地。对本病的概念、定义和病理变化等还有不同的认识,分类也较混乱。
心肌是心脏舒缩的动力结构,对物理性(如缺氧)、化学性(如药物、毒素)和生物性(如感染因子)等致病因素特别敏感。心肌的轻度损伤常表现为细胞核及细胞器的肥大,重度损伤表现为细胞结构的改建和细胞坏死,以及由此而导致的纤维化。这些变化既是各型心肌病的基本病变,又具有代偿适应意义。
一、原发性心肌病
1980年WHO将原发性心肌病分为三型:
(一)扩张性心肌病
扩张性(充血性)心肌病〔dilated (congestive)cardiomyopathy〕是原因不明的各种心肌疾病的最后结局,以心腔高度扩张和明显的心搏出力降低(心力衰竭)为特征。大多数病例可查出抗心内膜的自身抗体,其发病学意义尚不清楚。发病年龄为20~50岁,男多于女。患者多因两侧心力衰竭而就医。多数患者常因心力衰竭进行性加重而死亡或因心律心常而发生猝死。
【病变】
肉眼观,典型变化是两侧心室肥大,四个心腔扩张,心尖部变薄呈钝圆形(离心性肥大),状如牛心(图8-33)。重量比正常增加25%~50%(心重400~750g),由于心腔扩张,左心室壁厚度多在正常范围内;右心室壁常轻度增厚。心内膜纤维化在儿童患者较为明显,常伴有心内膜纤维弹性组织增生症。附壁血栓机化后可导致斑块状心内膜纤维化。由于左、右心室扩张,瓣环扩大,可导致二尖瓣及三尖瓣关闭不全。

图8-33 扩张性心肌病
两侧心室扩张,壁高度变薄(采自Becker和Anderson)
镜下,心肌细胞通常显示肥大和不同程度的伸长及肌浆变性,失去收缩成分。肥大的心肌细胞由于整个细胞的伸长,其横径多在正常范围,但其核大、浓染。心肌间质纤维化是此型心肌病最常见的变化,可见到血管周围和心肌细胞周围纤细的胶原纤维束,或致密的代替性纤维化灶。间质性纤维化通常以左心室心内膜、心肌为重。心内膜纤维化通常较轻,但附壁血栓处纤维化明显。此外,有些病例可见到淋巴细胞性间质性心肌炎,其特点是多发性淋巴细胞浸润灶伴有心肌细胞的变性和坏死。
(二)肥厚性心肌病
肥厚性心肌病(hypertrophic cardoiomyopathy)的特点是室间隔不匀称肥厚,心肌细胞异常肥大,排列方向紊乱以及收缩期二尖瓣帆向前移动等。肥厚的肌壁顺应性降低,致使心室充盈阻力增加。临床表现为不同程度的心室排空受阻而非充盈受限。根据左心室流出道有无梗阻现象可将其分为梗阻性和非梗阻性两型。右心室流出道或两心室流出道均受阻者少见。常导致猝死,亦可并发感染性心内膜炎。
【病变】
肉眼观,两侧心室显著肥大,心脏重量增加,为正常平均心重的1~2倍(成人患者平均心重582g,少数可达1000g)。绝大多数病例的室间隔厚度大于左室游离壁(图8-34,图8-35),肥厚可为局限性,可累及心基底部(主动脉瓣下)、室间隔中部或心尖区。收缩期二尖瓣向前移动与室间隔接触的结果,可导致二尖瓣增厚和主动脉瓣下心内膜纤维化。在心力衰竭发生之前,左心室一般不扩张。
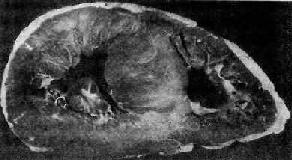
图8-34 肥厚性心肌病
图示心脏横切面(上面观),不对称性心室间隔肥厚(采自Becker和Anderson)
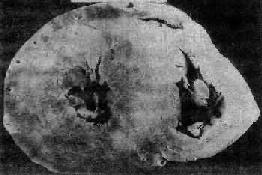
图8-35 肥厚性心肌病
心室横断面(上面观),左心室腔缩小,心室壁均匀增厚,整个心肌布满白色纤维化区,室间隔与后壁厚度之比仅稍大于正常值(1.4)
镜下,心肌细胞显著肥大,核大而浓染(图8-36),核周有亮区包围,组织化学染色证明为糖原堆积,具有一定的诊断意义。心肌细胞排列紊乱较其它型心肌病为甚,而且常呈旋涡状或缠绕呈簇状排列(图8-37),细胞内肌原纤维不呈平行排列,而是向各个方向、互相交错排列。常有间质纤维化灶形成,但以内膜纤维化,尤其位于主动脉瓣下区的内膜纤维化为突出。位于肥厚的室间隔内的冠状动脉分支管壁常有增厚现象。
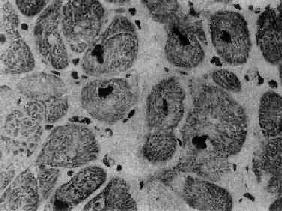
图8-36 肥厚性心肌病
心肌细胞肥大,核大浓染 (采自Edwards)

图8-37 肥厚性心肌病
心肌细胞排列紊乱 ×180(原放大)(采自Edwards)
电子显微镜观察,主要表现为相邻细胞的侧面出现细胞间连接的闰盘;发自某一肌原纤维Z带的肌丝可以不同角度插入另一肌原纤维的Z带,形成交织状排列。有时还可见肌丝从增宽的Z带向各个方向放射。但是,肌细胞侧对侧联接和肌原纤维排列紊乱并非肥厚性心肌病所独有的变化。
(三)限制性心肌病
限制性心肌病(restrictive cardiomyopathy)是以心室充盈受限制为特点。典型病变为心室内膜和内膜下心肌进行性纤维化,导致心室壁顺应性降低,心腔狭窄。因此亦称为心内膜心肌纤维化(endomyocardial fibrosis)。
【病变】
肉眼观,右心室内膜纤维化,尤以心尖部为明显,内膜增厚约2~3mm,灰白色,表面可有血栓形成。心尖部内膜纤维性增厚向上蔓延,可将乳头肌、肉柱埋陷在内,腱索变粗,缩短,可导致三瓣关闭不全。左心室内膜纤维化主要在流入道或心尖部,表面亦可有血栓形成。当二尖瓣后瓣叶与左心室后壁粘连时,引起二尖瓣关闭不全。
镜下,可见增厚的内膜主要为致密的玻璃样变的胶原纤维,可有钙化。表面可见陈旧的附壁血栓。心内膜下心肌常见萎缩、变性改变。
此外,嗜伊红细胞性心内膜心肌病(eosinophilic endomyocardial disease)可能是本型心肌病的亚型。浸润于心内膜心肌处的嗜伊红细胞脱颗粒,释出阳离子蛋白,可引起该处心肌的灶状坏死及纤维化。心室流入道及心尖部的附壁血栓和纤维化可导致心室限制性充盈障碍。
二、克山病
克山病(Keshan disease)是一种地方性心肌病(endemic cardiomyopathy)。1935年首先流行于黑龙江省克山县,当时对该病的本质认识不清,遂以此地名来命名,一直沿用至今。本病主要流行于我国东北、西北、华北及西南一带交通不便的山区或丘陵地带。病理学上以心肌的变性、坏死及修复后形成瘢痕为特点。临床上常有急性或慢性心功能不全表现。
【病因】
50年代以来对本病病因做了大量研究,但至今尚无定论。起初认为是一种地区流行性病毒性心肌炎,可能与柯萨奇B族病毒感染有关,但病毒分离和血清学检测未获规律性阳性结果。近年来发现病区粮食中硒含量明显低于非病区,患者的头发和血液中硒含量亦明显低于非病区人群。服用亚硒酸钠可控制一部分克山病的发作。1974~1977年间,我国在发病地区进行补硒的预防研究,结果表明,经补硒的儿童其克山病的发病例数显著低于对照组。应用硒制剂防治克山病在我国取得了一定的成果,然而,其它可能的致病因素尚有待进一步研究。
【病变】
本病的病变主要在心肌,可出现严重的变性、坏死及瘢痕形成。骨骼肌亦可有轻度变性或小灶状坏死。
肉眼观,心脏呈不同程度增大,两侧心室均扩张,致使心脏近于球形。重量亦增加,病程较长的慢性型病例心重量增加更甚,最重的可超过500g。心腔的扩张属肌原性,心壁变薄,乳头肌及肉柱变扁。少数病例在左心室肉柱间及左、右心耳内可有附壁血栓形成。若血栓脱落,可引起肺、肾、脾、脑等器官的栓塞和梗死。切面上见正常红褐色心肌内散布着数量不等的变性、坏死乃至瘢痕病灶。早期,坏死灶呈灰黄色,境界不清。瘢痕病灶呈灰白色、半透明,境界较清楚,呈星状或树枝状条纹,互相连接,有的呈较大的片块状或带状。心肌病变新旧交杂,色泽斑驳(图8-38)。
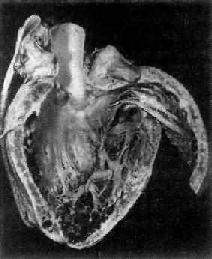
图8-38 克山病之心脏
左心室心肌有一些坏死灶及瘢痕形成,心室内可见附壁血栓
镜下,心肌细胞出现不同程度水肿,表现为胞浆内出现蛋白质颗粒(线粒体肿胀)和空泡变性。心肌坏死主要表现为凝固性坏死和液化性肌溶解。前者肌原纤维融合成均质红染物,核消失,继而坏死物通过细胞自身的及吞噬细胞的溶酶体酶溶解吸收,后者是在心肌空泡变性的基础上,肌原纤维及核发生酶性溶解液化,遗留肉膜空鞘。心肌坏死常呈灶状,病灶大小和形状不一(图8-39),呈散在分布,并多见于心肌内层,而且与冠状血管有密切关系,有的病变围绕冠状动脉分支呈袖套状分布(图8-40)。坏死灶最终被修复而成为瘢痕。

图8-39 克山病之心肌
心肌内可见多数坏死灶

图8-40 克山病之心肌坏死
坏死灶沿血管分布,坏死灶内肌纤维溶解消失
【临床表现】
根据患者发病缓急、病程长短及心肌代偿情况分为4型:
1.急性型 发病急骤,由于心肌病变比较广泛、严重,心肌收缩力明显减弱,心输出量在短时间内大幅度减少,重者出现心源性休克。由于供血不足,患者常有头昏、恶心、呕吐等症状。血压下降,心音弱,尤以第一心音减弱为著,并常有心律不齐。
2.亚急性型病情进展稍缓,心肌受损不如急性型那样严重,但心肌收缩力明显减弱。临床上出现明显的心力衰竭,特别是急性左心衰竭,有咳嗽、呼吸困难、满肺水泡音等征象。约经1~4周后,可发生全心衰竭,出现颈静脉怒张、肝肿大及全身水肿等。
3.慢性型 亦称痨型,病情发展缓慢,多由潜在型逐渐发展而成,少数由急性型或亚急性型转化而来。心脏代偿肥大,心腔扩张明显,临床上主要表现为慢性心功能不全。
4.潜在型 心脏受损较轻或因代偿功能较好,临床上多无明显的自觉症状。