第五节 肺疾病
一、慢性阻塞性肺病
慢性阻塞性肺病(chronic obstructive pulmonary diseases,COPD)是一种慢性气道阻塞性疾病的统称,主要指具有不可逆性气道阻塞的慢性支气管炎和肺气肿两种疾病。
(一)慢性支气管炎
慢性支气管炎(chronic bronchitis)是40岁以上男性人群中最常见的疾病之一,临床上以反复发作咳嗽、咳痰或伴有喘息症状为特征,且症状每年持续约3个月,连续两年以上。病情进展,常常并发肺气肿和慢性肺源性心脏病。是一种严重影响健康的慢性病。
【病因和发病机制】
慢性支气管炎往往是因多种因素长期综合作用所致。起病与感冒有密切关系,多在气候变化比较剧烈的季节发病。呼吸道反复病毒感染和继发性细菌感染是导致慢性支气管炎病变发展和疾病加重的重要原因。吸烟与慢性支气管炎的关系也是肯定的,吸烟者比不吸烟者的患病率高2~8倍,吸烟时间愈久,日吸烟量愈大,患病率愈高,戒烟可使病情减轻。此外,长期接触工业粉尘、大气污染和过敏因素也常是引起慢性支气管炎的原因,而机体抵抗力降低,呼吸系统防御功能受损则是发病的内在因素。
【病理变化】
各级支气管均可受累,受累的细支气管愈多,病变愈重,后果也愈严重。主要的病变有:①粘膜上皮纤毛倒伏,甚至脱失。上皮细胞变性、坏死脱落。上皮再生时,杯状细胞增多,并可发生鳞状上皮化生(图9-10);②粘液腺肥大、增生,分泌亢进,浆液腺发生粘液化(图9-11);③管壁充血,淋巴细胞、浆细胞浸润;④管壁平滑肌束断裂、萎缩,而喘息型患者,平滑肌束可增生、肥大,管腔变窄;⑤软骨可发生变性、萎缩,钙化或骨化。支气管炎反复发作的结果,病变不仅逐渐加重,而且逐级向纵深发展蔓延,受累的细支气管数量也不断增多。细支气管因管壁薄,炎症易向管壁周围组织及肺泡扩展,导致细支气管周围炎,而且还可发生纤维闭塞性细支气管炎,是引起慢性阻塞性肺气肿的病变基础。

图9-10 慢性支气管炎
支气管粘膜纤毛上皮出现较多杯状细胞,部分呈磷状上皮化生

图9-11 慢性支气管炎
支气管粘膜上皮出现较多杯状细胞,固有层及粘膜下层内有慢性炎性细胞浸润,腺体呈粘液化
【临床病理联系】
患者因支气管粘膜的炎症和分泌物增多,而出现咳嗽、咳痰症状。痰一般呈白色粘液泡沫状。在急性发作期,咳嗽加重,并出现粘液脓性或脓性痰。由于支气管痉挛或支气管狭窄及粘液、渗出物阻塞而引起喘息。检查时,两肺可闻及哮鸣音、干湿啰音。有的患者因粘膜和腺体萎缩(慢性萎缩性支气管炎),分泌物减少,痰量减少甚或无痰。病变导致小气道狭窄或阻塞时,出现阻塞性通气障碍,表现为第1秒用力呼吸量和最大通气量明显降低,合併肺气肿时,肺残气量明显增多,肺总量也增大。
(二)肺气肿
肺气肿(pulmonary emphysema)是指呼吸细支气管以远的末梢肺组织因残气量增多而呈持久性扩张,并伴有肺泡间隔破坏,以致肺组织弹性减弱,容积增大的一种病理状态。在成人尸检例中,约50%可发现不同程度的肺气肿,其中约6.5%的患得因此病死亡。
【病因和发病机制】
肺气肿是支气管和肺疾病常见的併发症。与吸烟、空气污染、小气道感染、尘肺等关系密切,尤其是慢性阻塞性细支气管炎是引起肺气肿的重要原因。发病机制与下列因素有关:
1.阻塞性 通气障碍慢性细支气管炎时,由于小气道的狭窄、阻塞或塌陷,导致了阻塞性通气障碍,使肺泡内残气量增多,而且,细支气管周围的炎症,使肺泡壁破坏、弹性减弱,更影响到肺的排气能力,末梢肺组织则因残气量不断增多而发生扩张,肺泡孔扩大,肺泡间隔也断裂,扩张的肺泡互相融合形成气肿囊腔。
此外,细支气闭塞时,吸入的空气可经存在于细支气管和肺泡之间的Lambert孔进入闭塞远端的肺泡内(即肺泡侧流通气),而呼气时,Lambert孔闭合,空气不能排出,也是导致肺泡内储气量增多、肺泡内压增高的因素。
2.弹性蛋白酶增多、活性增高与肺气肿发生有关的内源性蛋白酶主要是中性粒细胞和单核细胞释放的弹性蛋白酶。此酶能降解肺组织中的弹性硬蛋白、结缔组织基质中的胶原和蛋白多糖,破坏肺泡壁结构。慢性支气管炎伴有肺感染、尤其是吸烟者,肺组织内渗出的中性粒细胞和单核细胞较多,可释放多量弹性蛋白酶。同时,中性粒细胞和单核细胞还可生成大量氧自由基,能氧化α1-抗胰蛋白酶活性中心的蛋氨酸使之失活。α1-抗胰蛋白酶乃弹性蛋白酶的抑制物,失活后则增强了弹性蛋白酶的损伤作用。
α1-抗胰蛋白酶由肝细胞产生,是一种分子量为45000~56000的糖蛋白,它能抑制蛋白酶、弹性蛋白酶、胶原酶等多种水解酶的活性。遗传性α1-抗胰蛋白酶缺乏是引起原发性肺气肿的原因,α1-抗胰蛋白酶缺乏的家族,肺气肿的发病率比一般人高15倍,主要是全腺泡型肺气肿。但是,在我国因遗传性α1-抗胰蛋白酶缺乏引起的原发性肺气肿非常罕见,并不重要。而最重要的也是最常见的是慢性阻塞性肺气肿(继发性肺气肿)。
【类型及其病变特点】
肺气肿病变发生在肺腺泡(acinus),即Ⅰ级呼吸细支气管所分布的肺组织范围内。属肺泡性肺气肿(alveolar emphysema)。根据病变的确切解剖部位及分布范围的不同可分为:
1.弥漫性肺气肿
(1)腺泡中央型肺气肿:腺泡中央型肺气肿(centriacinar emphysema)病变累及肺腺泡的中央部分,呼吸细支气管病变最明显,呈囊状扩张(图9-12)。而肺泡管、肺泡囊变化则不明显。
(2)全腺泡型肺气肿:全腺泡型肺气肿(panacinar emphysema)病变累及肺腺泡的各个部位,从终末呼吸细支气管直至肺泡囊和肺泡均呈弥漫性扩张,遍布于肺小叶内(图9-13)。如果肺泡间隔破坏较严重,气肿囊腔可融合成直径超过1cm的大囊泡,形成大泡性肺气肿(图9-14)。
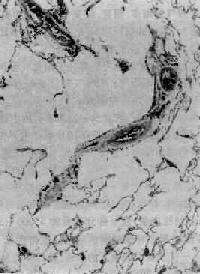
图9-12 腺泡中央型肺气肿
呼吸细支气管呈囊状扩张,伴行肺动脉(径80μm)管壁增厚,其分支内膜增厚,管腔极度狭窄

图9-13 全腺泡型肺气肿
末梢呼吸道弥漫性扩张,呈小囊状遍布于肺小叶内(径>200μm)
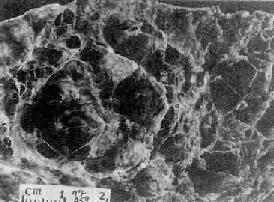
图9-14 左肺下叶大泡性肺气肿
在全腺泡型肺气肿基础上,胖有直径超过1cm的大囊泡
(3)腺泡周围型肺气肿:腺泡周围型肺气肿(periacinar emphysema)也称隔旁肺气肿(paraseptal emphysema),病变主要累及肺腺泡远端部位的肺泡囊,而近端部位的呼吸细支气管和肺泡管基本正常。常合并有腺泡中央型和全腺泡型肺气肿。
2.局限性肺气肿
(1)不规则型肺气肿:不规则型肺气肿(irregular emphysema)也称瘢痕旁肺气肿(paracicatricial emphysema),病变主要发生在瘢痕附近的肺组织,肺腺泡不规则受累,确切部位不定,一般是发生在呼吸细支气管远侧端,肺泡囊有时也受累。
(2)肺大泡:肺大泡(bullae of lung)病变特点是局灶性肺泡破坏,小叶间隔也遭破坏,往往形成直径超过2cm的大囊泡,常为单个孤立位于脏层胸膜下。而其余肺结构可正常。
间质性肺气肿(interstitial emphysema)是由于肺泡壁或细支气壁破裂,气体逸入肺间质内,在小叶间隔与肺膜连接处形成串珠状小气泡,呈网状分布于肺膜下。
【病理变化】
肉眼观:气肿肺显著膨大,边缘钝圆,色泽灰白,表面常可见肋骨压痕,肺组织柔软而弹性差,指压后的压痕不易消退,触之捻发音增强。
镜下:肺泡扩张,间隔变窄,肺泡孔扩大,肺泡间隔断裂,扩张的肺泡融合成较大的囊腔。肺毛细血管床明显减少,肺小动脉内膜呈纤维性增厚。小支气管和细支气管可见慢性炎症。腺泡中央型肺气肿的气肿囊泡为扩张的呼吸细支气管,在近端囊壁上常可见呼吸上皮(柱状或低柱状上皮)及平滑肌束的残迹。全腺泡型肺气肿的气肿囊泡主要是扩张变圆的肺泡管和肺泡囊,有时还可见到囊泡壁上残留的平滑肌束片断,在较大的气肿囊腔内有时还可见含有小血管的悬樑。
【临床病理联系】
肺气肿患者的主要症状是气短,轻者仅在体力劳动时发生,随着气肿程度加重,气短逐渐明显,甚至休息时也出现呼吸困难,并常感胸闷。每当合并呼吸道感染时,症状加重,并可出现缺氧、酸中毒等一系列症状。肺功能检查诊断肺气肿的标准是残气量超过肺总量的35%,最大通气量低于预计值的80%,肺总量超过预计值的100%,1秒用力呼吸量低于肺活量的60%。典型肺气中患者的胸廓前后径增大,呈桶状胸。胸廓呼吸运动减弱。叩诊呈过清音,心浊音界缩小或消失,肝浊音界下降。语音震颤减弱。听诊时呼吸音减弱,呼气延长,用力呼吸时两肺底部可闻及湿啰音和散在的干啰音。剑突下心音增强,肺动脉瓣第二音亢进。
肺气肿的严重后果有①肺源性心脏病及衰竭。②肺大泡破裂后引起自发性气胸,并可导致大面积肺萎陷。③呼吸衰竭及肺性脑病。由于外呼吸功能严重障碍,导致动脉血PaO2<8kPa(60mmHg),伴有或不伴有PaCO2<6.67kPa(50mmHg)的病理过程,称为呼吸衰竭(respiratory failure)。呼吸衰竭时发生的低氧血症和高碳酸血症会引起各系统的代谢功能严重紊乱。中枢神经系统对缺氧最为敏感,愈是高级部位敏感性愈高。随着缺氧程度的加重,可出现一系列中枢神系统功能障碍,由开始的大脑皮层兴奋性增高而后转入抑制状态。病人表现由烦燥不安、视力和智力的轻度减退,逐渐发展为定向和记忆障碍,精神错乱,嗜睡,惊厥以至意识丧失。迅速发生的CO2潴留也能引起中枢神经功能障碍,病人常出现头痛、头晕、烦燥不安、言语不清、扑翼样震颤、精神错乱以及嗜睡、昏迷、呼吸抑制等“二氧化碳麻醉”症状。由呼吸衰竭造成的以脑功能障碍为主要表现的综合征,称为肺性脑病(pulmonary encephalopathy),可能是由于低氧血症、高碳酸血症,以及酸碱平衡紊乱导致神经细胞变性、坏死和脑血液循环障碍引起脑血管扩张、脑水肿、灶性出血、颅内压升高甚至脑疝形成等因素综合作用所致。
二、肺炎
肺炎(pneumonia)通常是指肺的急性渗出性炎症,为呼吸系统的多发病、常见病。据世界卫生组织调查,肺炎死亡率占呼吸系统急性感染死亡率的75%。在我国,各种致死病因中,肺炎占第5位。肺炎可由不同的致病因子引起,根据病因可将肺炎分为感染性(如细菌性、病毒性、支原体性、真菌性和寄生虫性)肺炎,理化性(如放谢性、吸入性的类脂性)肺炎以及变态反应性(如过敏性和风湿性)肺炎。由于致病因子和机体反应性的不同,炎症发生的部位、累及范围和病变性质也往往不同。炎症发生于肺泡内者称肺泡性肺炎(大多数肺炎为肺泡性),累及肺间质者称间质性肺炎。病变范围以肺小叶为单位者称小叶性肺炎,累及肺段者称节段性肺炎,波及整个或多个大叶者称大叶性肺炎。按病变性质可分为浆液性、纤维素性、化脓性、出血性、干酪性、肉芽肿性或机化性肺炎等不同类型。
(一)细菌性肺炎
1.大叶性肺炎 大叶性肺炎(lobar pneumonia)主要是由肺炎链球菌感染引起,病变起始于肺泡,并迅速扩展至整个或多个大叶的肺的纤维素性炎。多见于青壮年,临床表现为骤然起病、寒战高烧、胸痛、咳嗽、吐铁锈色痰、呼吸困难,并有肺实变体征及白细胞增高等。大约经5~10天,体温下降,症状消退。
【病因和发病机制】
95%以上的大叶性肺炎由肺炎链球菌引起,尤以Ⅲ型者毒力最强。此外,肺炎杆菌、金黄色葡萄球菌、溶血性链球菌、流感嗜血杆菌也可引起。受寒、疲劳、醉酒、感冒、麻醉、糖尿病、肝、肾疾病等均可为肺炎的诱因。此时,呼吸道的防御功能被削弱,机体抵抗力降低,易发生细菌感染。细菌侵入肺泡后在其中繁殖,特别是形成的浆液性渗出物又有利于细菌繁殖,并使细菌通过肺泡间孔或呼吸细支气管迅速向邻近肺组织蔓延,从而波及整个大叶,在大叶之间的蔓延则系带菌渗出液经叶支气管播散所致。
【病理变化】
病变一般发生在单侧肺,多见于左肺下叶,也可同时或先后发生于两个以上肺叶。病变基本特征是肺的微循环障碍。由于毛细血管通透性增高,大量纤维蛋白原渗出于肺泡,使肺组织大面积广泛实变。病变早期,肺叶充血、水肿,肺泡腔内有大量浆液性渗出物,混有少数红细胞、中性粒细胞和巨噬细胞,并含有大量细菌。1~2天后,即有大量纤维蛋白原渗出,肺泡腔内充满混有红细胞、中性粒细胞、巨噬细胞的纤维素性渗出物,纤维素丝可穿过肺泡间孔与相邻肺泡中的纤维素网相连(图9-15)。病变肺叶质实如肝,明显肿胀,重量增加,呈灰白色(图9-16)。如血管损伤较重、出血较多,外观可呈红色。大约经5~10天,炎症消退,细菌被吞噬细胞吞噬清除,渗出物被溶解,或经淋巴管吸收或被咳出。大叶性肺炎时,肺组织常无坏死,肺泡壁结构也未遭破坏,愈复后,肺组织可完全恢复其正常结构和功能。
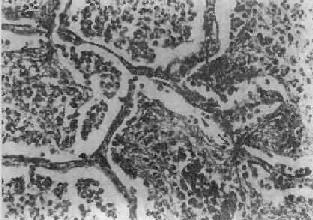
图9-15 大叶性肺炎
肺泡腔内充满纤维素性渗出物,纤维素丝穿过肺泡间孔,使相邻肺泡内的纤维素网互相连接

图9-16 大叶性肺炎
左下叶实变,呈灰白色,肺叶明显肿胀
【合并症】
(1)肺肉质变:因吞噬细胞数量少或功能缺陷,渗出物不能被完全吸收清除时,则由肉芽组织予以机化(图9-17),病变部位肺组织变成褐色肉样纤维组织,称肉质变(carnification)。
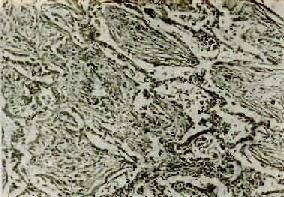
图9-17 肺肉质变
肺泡腔内炎性渗出物已被结缔组织所替代
(2)肺脓肿及脓胸或脓气胸:多见于由金黄色葡萄球菌引起的肺炎。
(3)纤维素性胸膜炎:是肺内炎症直接侵犯胸膜的结果。
(4)败血症或脓毒败血症:见于严重感染时,细菌侵入血流繁殖所致。
(5)感染性休克:严重的肺炎链球菌或金黄色葡萄球菌感染引起严重的毒血症时可发生休克,称休克型或中毒性肺炎,病死率较高。
【临床病理联系】
疾病发展过程中病变表现不一,临床体征也不相同。疾病早期时,主要病变是肺泡腔内浆液渗出,听诊可闻湿啰音,X线检查仅见肺纹理增深。肺实变时,由于肺泡膜面积减少,可出现肺泡通气和血流比例失调而影响换气功能,使肺静脉血不能充分氧合,患者乃出现紫绀或呼吸困难。渗出物中红细胞为肺泡巨噬细胞吞噬,崩解后形成含铁血黄素混入痰中,使痰呈铁锈色。痰中可检出“心衰细胞”。肺实变的体征是,肺泡呼吸音减弱或消失,出现支气管呼吸音,语音震颤增强,叩诊呈浊音。因常并发纤维素性胸膜炎,患者有胸痛,听诊可闻胸膜摩擦音。X线检查,可见大叶性或段性分布的均匀性密度增高阴影。病变消散时,渗出物溶解液化,肺部可闻及捻发音,X线表现为散在不均匀片状阴影,约在2~3周后阴影方完全消散。抗生素治疗,可缩短病程,减轻病变,合并症也大为减少。
2.小叶性肺炎 小叶性肺炎(lobular pneumonia)主要由化脓菌感染引起,病变起始于细支气管,并向周围或末梢肺组织发展,形成以肺小叶为单位、呈灶状散布的肺化脓性炎。因其病变以支气管为中心故又称支气管肺炎(bronchopneumonia)。主要发生于小儿和年老体弱者。
【病因和发病机制】
小叶性肺炎主要由细菌感染引起,常见的致病菌有葡萄球菌、链球菌、肺炎球菌、流感嗜血杆菌、绿脓杆菌和大肠杆菌等。这些细菌通常是口腔或上呼吸道内致病力较弱的常驻寄生菌,往往在某些诱因影响下,如患传染病、营养不良、恶病质、慢性心力衰竭、昏迷、麻醉、手术后等,使机体抵抗力下降,呼吸系统的防御功能受损,细菌得以入侵、繁殖,发挥致病作用,引起支气管肺炎。因此,支气管肺炎常是某些疾病的并发症,如所谓麻疹后肺炎、手术后肺炎、吸入性肺炎、坠积性肺炎等等。
【病理变化】
小叶性肺炎的病变特征是肺组织内散布一些以细支气管为中心的化脓性炎症病灶。常散布于两肺各叶,尤以背侧和下叶病灶较多。病灶大小不等,直径多在1cm左右(相当于肺小叶范围),形状不规则,色暗红或带黄色(图9-18)。严重者,病灶互相融合甚或累及全叶,形成融合性支气管肺炎(confluent bronchopneumonia)。镜下,病灶中支气管、细支气及其周围的肺泡腔内流满脓性渗出物,纤维蛋白一般较少(图9-19)。病灶周围肺组织充血,可有浆液渗出、肺泡过度扩张等变化。由于病变发展阶段的不同,各病灶的病变表现和严重程度亦不一致。有些病灶完全化脓,支气管和肺组织遭破坏,而另一些病灶内则可仅见浆液性渗出,有的还停留于细支气管及其周围炎阶段。

图9-18 支气管肺炎
图中大小不等的不规则形区域即实变的支气管肺炎病灶
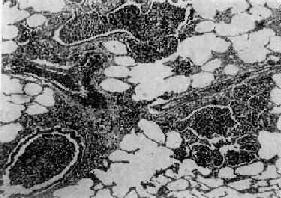
图9-19 支气管肺炎
图中见灶状实变的肺组织,肺泡内充满以中性粒细胞为主的炎性渗出物;病灶中有发炎的细支气管
【并发症】
小叶性肺炎发生并发症的危险性比大叶性肺炎大得多。可并发心力衰竭、呼吸衰竭、脓毒败血症、肺脓肿及脓胸等。支气管破坏较重且病程较长者,可导致支气管扩张。
【临床病理联系】
因小叶性肺炎多为其他疾病的并发症,其临床症状常为原发性疾病所掩盖。由于支气管粘膜的炎症刺激而引起咳嗽,痰呈粘液脓性。因病变常呈灶性散布,肺实变体征一般不明显。病变区细支管和肺泡内含有渗出物,听诊可闻湿啰音。X线检查,可见肺野内散在不规则小片状或斑点状模糊阴影。本病发现及时,治疗得当,肺内渗出物可完全吸收而痊愈。但在幼儿,年老体弱者,特别是并发于其他严重疾病时,预后大多不良。
(二)病毒性肺炎
病毒性肺炎(viral pneumonia)常常是因上呼吸道病毒感染向下蔓延所致。患者多为儿童,症状轻、重不等,但婴幼儿和老年患者病情较重。一般多为散发,偶可酿成流行。引起肺炎的病毒种类较多,常见的是流感病毒、还有呼吸道合胞病毒、腺病毒、副流感病毒、麻疹病毒、巨细胞病毒等等,也可由一种以上病毒混合感染并可继发细菌感染。病毒性肺炎的病情、病变类型及其严重程度常有很大差别。
【病理变化】
早期或轻型病毒性肺炎表现为间质性肺炎,炎症从支气管、细支气管开始,沿肺间质发展,支气管、细支气管壁及其周围、小叶间隔以及肺泡壁等肺间质充血、水肿,有一些淋巴细胞和单核细胞浸润,肺泡壁明显增宽(图9-20)。肺泡腔内一般无渗出物或仅有少量浆液。病变较重者,肺泡也可受累,出现由浆液、少量纤维蛋白、红细胞及巨噬细胞组成的炎性渗出物,甚至可发生组织坏死。有些病毒性肺炎(如流感病毒肺炎,麻疹病毒肺炎、腺病毒肺炎等)肺泡腔内渗出较明显,渗出物浓缩凝结成一层红染的膜样物贴附于肺泡内表面,即透明膜形成。支气管上皮的肺泡上皮也可增生,甚至形成多核巨细胞。麻疹病毒肺炎的病变特点为在间质性肺炎的基础上,肺泡壁上有透明膜形成,并有较多的多核巨细胞(巨细胞肺炎),在增生的上皮细胞和多核巨细胞的胞浆内和胞核内可检见病毒包含体。病毒包含体常呈球形,约红细胞大小,呈嗜酸性染色,均质或细颗粒状,其周围常有一清晰的透明晕。其他一些病毒性肺炎也可在增生的支气管上皮、支气管粘液腺上皮或肺泡上皮细胞内检见病毒包含体。如腺病毒肺炎可在增生的上皮细胞核内(图9-21),呼吸道合胞病毒肺炎可在增生的上皮细胞胞浆内,巨细胞病毒肺炎也可在增生的上皮细胞核内检见病毒包含体。检见包含体是病理组织学诊断病毒性肺炎的重要依据。
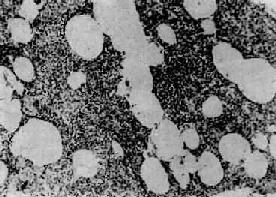
图9-20 间质性肺炎
肺泡壁及细支气管周围肺间质内有大量炎性细胞(主为单核细胞)浸润。肺泡壁明显增宽。肺泡腔内无渗出物
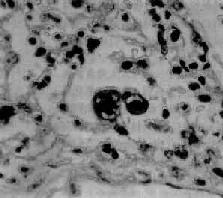
图9-21 腺病毒肺炎
图中央可见肿大肺泡上皮细胞中的核内包含体
有些混合感染,如麻疹病毒合并腺病毒感染,特别是又继发细菌感染的病毒性肺炎,病变更为严重,肺炎病灶可呈小叶性、节段性或大叶性分布。支气管和肺组织明显坏死、出血,并可混杂化脓性病变,从而掩盖了病毒性肺炎原来的病变特征。
(三)支原体肺炎
支原体肺炎(mycoplasmal pneumonia)是由肺炎支原体(mycoplasma pneumoniae)引起的一种间质性肺炎。支原体系介于细菌和病毒之间的微生物,共有30余种,其中多种可寄生于人体,但不致病,仅有肺炎支原体能引起呼吸道疾病。各种肺炎中约有5%~10%乃由肺炎支原体引起。主要经飞沫感染,秋、冬季节发病较多,儿童和青年发病率较高,通常为散发性,偶尔流行。患者起病较急,多有发热、头痛、咽痛及剧烈咳嗽(常为干性呛咳)等症状。胸部检查,可闻干、湿啰音。X线检查,肺部呈段性分布的纹理增加及网织状阴影。白细胞计数有轻度升高,淋巴细胞和单核细胞增多,痰、鼻分泌物及咽喉拭子能培养出肺炎支原体。
【病理变化】
肺炎支原体感染可引起整个呼吸道的炎症。肺部病变常仅累及一个肺叶,以下叶多见。病变主要发生于肺间质,病灶呈段性分布,暗红色,切面可有少量红色泡沫状液体溢出。气管或支气管腔内也可见粘液性渗出物。胸膜光滑。镜下,病变区域肺泡间隔明显增宽,有大量淋巴细胞、浆细胞和单核细胞浸润,肺泡腔内无渗出物或仅有少量混有单核细胞的浆液性渗出液。小支气管和细支气管壁及其周围组织也常有炎性细胞浸润。重症病例,上皮亦可坏死脱落,往往伴有中性粒细胞浸润。支原体肺炎预后良好。死亡率在0.1%~1%之间。
三、职业性肺疾病
在职业活动、特别是生产过程中,因长期吸入有害粉尘,引起以肺广泛纤维化为主要病变的疾病,统称尘肺(pneumoconiosis)。尘肺是我国一种法定职业病。属职业性尘肺的病种较多,按粉尘的化学性质可将其分为无机尘肺和有机尘肺两大类。无机尘肺中常见的有硅肺、煤工尘肺、石棉肺等。有机尘肺是因吸入各种有机尘埃,最常见的是由霉菌的代谢产物或动物性蛋白质引起的尘肺,如农民肺、蔗尘肺、蘑菇肺、麦芽肺和饲禽者肺等。尘肺对健康危害极大,关键在于预防。改革不合理的生产过程,建立粉尘监测制度,切实落实综合防尘措施。不接触粉尘或减少吸入粉尘的机会,对于粉尘作业工人定期体检,做到早期检查、早期诊断,对已确诊为尘肺患者及早调离粉尘作业,并进行必要的治疗,完全可以控制和减少尘肺的发病率。
(一)矽肺
矽肺(silicosis)是在生产环境中长期吸入大量含游离二氧化硅(SiO2)粉尘微粒所引起的以肺纤维化为主要病变的全身性疾病。游离二氧化硅主要存在于石英中,石英成分中SiO2占97%~99%。约有70%的矿石中均含有较多的SiO2。长期从事开矿、采石作业、坑道作业以及在石英粉厂、玻璃厂、耐火材料厂、陶瓷厂和搪瓷厂生产作业的工人易患本病。矽肺是危害最严重的一种职业病,其特点是发展缓慢,即使在脱离矽尘作业后,病变仍然继续缓慢发展。患者多在接触矽尘10~15年后才发病。若因吸入高浓度、高游离二氧化硅含量的矽尘,经1~2年后发病者,称速发型矽肺。矽肺的早期即有肺功能损害,但因肺的代偿能力很强,患者往往无症状,随着病变的发展,尤其是合并肺结核和肺心病时,则逐渐出现不同程度的呼吸和心功能障碍。
【病因和发病机制】
游离二氧化硅是矽肺的致病因子。矽肺的发生、发展与矽尘中游离二氧化硅的含量,生产环境中矽尘的浓度、分散度,从事矽尘作业的工龄及机体防御功能等因素有关。矽尘粒子愈小,分散度愈度,在空气中的沉降速度愈慢,被吸入的机会就愈多,致病作用亦愈强。一般来说,大于5μm的矽尘往往被阻留在上呼吸道,并可被呼吸道的防御装置清除。小于5μm的矽尘才能被吸入肺泡,并进入肺泡间隔,引起病变。尤以1~2μm的矽尘微粒引起的病变最为严重。
吸入肺泡内的矽尘微粒被肺巨噬细胞吞噬,沿肺淋巴流经细支气管周围、小血管周围、小叶间隔和胸膜再到达肺门淋巴结。当淋巴道阻塞后,矽尘沉积于肺间质内引起矽肺病变。若局部沉积的矽尘量多,引起肺巨噬细胞局灶性聚积,可导致矽结节形成;若矽尘散在分布,则引起弥漫性肺间质纤维化。矽肺的发病机制尚未完全阐明。一般认为,矽尘被肺巨噬细胞吞噬后,在游离二氧化硅的毒性作用下,巨噬细胞大量死亡崩解或发生功能和生物学行为改变,释放出一些致纤维化因子、包括巨噬细胞生长因子(MDGF),白细胞介素Ⅰ(IL-1)和纤维连结蛋白(FN)等,促进成纤维细胞增生和胶原形成,导致纤维化。至于巨噬细胞死亡的原因,主要是由于矽尘被巨噬细胞吞噬后,存在于次级溶酶体中,矽尘表层中的SiO2逐渐与水聚合成硅酸(系一种强的成氢键化合物),其羟基基团与溶酶体膜脂蛋白结构上的受氢原子(氧、氮或硫)间形成氢键,改变了溶酶体膜的脂质分子构型,从而破坏了膜的稳定性或完整性。溶酶体膜通透性增高或破裂,其中所含的大量水解酶溢出到细胞内,导致巨噬细胞自溶崩解。巨噬细胞死亡崩解后,释出的矽尘又被其它巨噬细胞吞噬,如此反复进行,使病变不断发展、加重。这也可解释何以患者脱离矽尘作业后肺部病变仍然会继续发展的缘由。
随着免疫学的发展,大量关于矽肺免疫的研究表明,在矽肺发生、发展过程中,有免疫因素参与。根据对矽结节玻璃样变组织的生化分析,其中球蛋白含量明显高于胶原含量,而有别于一般的玻璃样变组织的成分。动物实验证明,矽肺病变的纤维化程度与浆细胞反应强度呈正相关,提示矽肺的纤维化与抗原抗体反应有关。用荧光免疫组织化学方法观察矽结节,发现在胶原纤维及其间隙中有大量γ-球蛋白沉积,主要是IgG和IgM。如将尸检取得的矽结节玻璃样变组织制成匀浆,给家兔注射后,能产生抗人γ-球蛋白抗体。有人认为,浆细胞产生的免疫球蛋白通过形成抗原抗体复合物参与矽肺的发病。对矽肺患者作体液免疫测定发现,血清中IgG和IgM浓度增高,抗肺自身抗体、抗核抗体和类风湿因子检出率也较高。但关于矽肺免疫的抗原物质目前还未提取出来,多认为有3种可能性:①矽尘作为半抗原与机体的蛋白质结合构成复合抗原;②矽尘表面吸附的γ-球蛋白转化为自身抗原;③矽尘导致巨噬细胞死亡崩解后释放自身抗原。现已有很多证据表明,巨噬细胞死亡崩解后释放抗原的可能性最大。总之,矽肺的病因是明确的,发病机制极为复杂,在发病过程中可能有多种因素参与,它们互相影响、互为因果,共同促进矽肺的发生和发展。
【病理变化】
矽肺的基本病变是肺组织内矽结节形成和弥漫性间质纤维化。矽结节是矽肺的特征性病变,结节境界清楚,直径2~5mm,呈圆形或椭圆形,灰白色,质硬,触之有砂样感。随着病变的发展,结节可融合成团块状,在团块的中央,由于缺血、缺氧而发生坏死、液化,形成矽肺性空洞(silicotic cavity)。矽结节的形成过程大致分为三个阶段:①细胞性结节,由吞噬矽尘的巨噬结胞局灶性聚积而成,巨噬细胞间有网状纤维,这是早期的矽结节;②纤维性结节,由纤维母细胞、纤维细胞和胶原纤维构成;③玻璃样结节,玻璃样变从结节中央开始,逐渐向周围发展,往往在发生玻璃样变的结节周围又有新的纤维组织包绕。镜下,典型的矽结节是由呈同心圆状或旋涡状排列的、已发生玻璃样变的胶原纤维构成(图9-22)。结节中央往往可见内膜增厚的血管。用偏光显微镜观察,可以发现沉积在矽结节和肺组织内呈双屈光性的矽尘微粒。除矽结节外,肺内还有不同程度的弥漫性间质纤维化(图9-23),范围可达全肺2/3以上。此外,胸膜也因纤维组织弥漫增生而广泛增厚,在胸壁上也可形成胸膜胼胝,甚至可厚达1~2cm。肺门淋巴结内也有矽结节形成和弥漫性纤维化及钙化,淋巴结因而肿大、变硬。
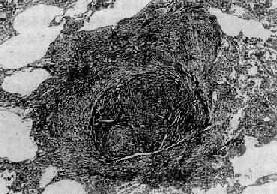
图9-22 矽肺
矽结节,由呈旋涡状排列的已发生玻璃样变的胶原纤维构成

图9-23 矽肺
肺组织呈弥漫性纤维化
矽尘沿血道转运,可在肝、脾、骨髓等处形成矽结节。
矽肺的分期和病变特征
根据肺内矽结节的数量、分布范围和直径大小,可将矽肺分为三期:
Ⅰ期矽肺:矽结节主要局限在淋巴系统。肺组织中矽结节数量较少,直径一般在1~3mm,主要分布在两肺中、下叶近肺门处。X线检查,肺野内可见一定数量的类圆形或不规则形小阴影,其分布范围不少于两个肺区。此时,肺的重量、体积和硬度无明显改变。胸膜上可有矽结节形成,但胸膜增厚不明显。
Ⅱ期矽肺:矽结节数量增多、体积增大,可散于全肺,但仍以肺门周围中、下肺叶较密集,总的病变范围不超过全肺的1/3。X线表现为肺野内有较多量直径不超过1cm的小阴影,分布范围不少于四个肺区。此时,肺的重量、体积和硬度均有增加,胸膜也增厚。
Ⅲ期矽肺(重症矽肺):矽结节密集融合成块。X线表现有大阴影出现,其长径不小于2cm,宽径不小于1cm,此时,肺的重量和硬度明显增加。解剖取出新鲜肺标本可竖立不倒(图9-24),切开时阻力甚大,并有砂粒感。浮沉试验,全肺入水下沉。团块状结节的中央可有矽肺空洞形成。结节之间的肺组织常有明显的灶周肺气肿,有时肺表面还可见到肺大泡。
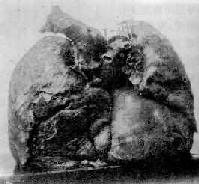
图9-24 Ⅲ期矽肺并发肺心病
两肺坚实能竖立,胸膜增厚。右心显著肥大、扩大,心尖由右心构成
【合并症】
1.矽肺结核病 矽肺合并结核病时称为矽肺结核病(silicotuberculosis)。愈是晚期、重症矽肺,肺结核的合并率愈高。Ⅲ期矽肺的合并率达60%~70%或更高。矽肺患者易合并肺结核可能是因游离二氧化硅对巨噬细胞的毒性损害以及肺间质弥漫性纤维化,导致肺的血液循环和淋巴循环障碍,从而降低了肺组织对结核杆菌的防御能力之故。矽肺结核病时,矽肺病变和结核病变可分开存在,也可混合存在(9-25)。矽肺结核病的病变比单纯矽肺和单纯肺结核的病变发展更快,累及范围更广,更易形成空洞。矽肺结核性空洞的特点是数目多,直径大,空洞壁极不规则。较大的血管易被侵蚀,可导致患者大咯血死亡。
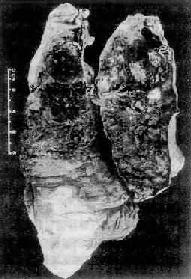
图9-25 矽肺结核病
图中可见胸膜增厚,切面上可见矽肺引起的肺气肿小囊泡。右侧白色斑块为结核病灶
2.肺感染由于矽肺患者抵抗力低,又有慢性阻塞性肺疾病,小气道引流不畅,故易继发细菌或病毒感染。尤其在有弥漫性肺气肿的情况下,肺感染可诱发呼吸衰竭而致死。
3.慢性肺源性心脏病 约有60%~75%的矽肺患者并发肺心病。这是因为肺间质弥漫性纤维化,肺毛细血管床减少,肺循环阻力增加。同时,矽结节内小血管常因闭塞性血管内膜炎,管壁纤维化,使管腔狭窄乃至闭塞,血管也扭曲、变形,尤以肺小动脉的损害更为明显,加之因呼吸功能障碍造成的缺氧,引起肺小动脉痉挛,均可导致肺循环阻力增加、肺动脉高压和右心室肌壁肥厚,心腔扩张。重症患者可因右心衰竭而死亡。
4.肺气肿和自发性气胸晚期矽肺患者常有不同程度的弥漫性肺气肿,主要是阻塞性肺气肿。有时,在脏层胸膜下还可出现肺大泡。气肿囊腔破裂引起自发性气胸。
(二)石棉肺
石棉肺(asbestosis)是因长期吸入石棉粉尘引起的以肺间质纤维化和胸膜肥厚为主要病变的疾病。石棉是一种天然的矿物结晶,其化学成分是含有铁、镁、铝、钙、镍等元素的硅酸盐复合物。石棉纤维具有耐酸、碱,隔热,绝缘等特性,在工业上用途很广。石棉矿的开采、选矿、运输工,石棉加工厂的分类、弹棉工,石棉制品的绝缘、隔热材料制品工等都可因长期吸入石棉粉尘而发生职业性石棉肺。发病工龄一般在10年左右。临床上,患者主要出现慢性支气管炎、肺气肿的症状,如咳嗽、咳痰、气急、胸胀痛等,晚期出现肺功能障碍和肺心病的症状和体征,患者痰内可检见石棉小体。
【发病机制】
吸入的石棉粉尘多数停留在呼吸性细支气管,仅有部分抵达肺泡,进入肺间质,有的还可被运输到脏层胸膜,引起肺间质和胸膜的结缔组织增生。至于纤维化形成的机制还不太清楚,可能主要是由于石棉纤维直接刺激纤维母细胞,促使脯氨酸羟化为羟脯氨酸,加速胶原合成,因而形成纤维化。除直接刺激作用外,也可能与石棉对巨噬细胞的毒性损害有关。虽然石棉对巨噬细胞的毒性作用比二氧化硅小,但石棉中的Mg2+对细胞膜也有溶解损伤作用。此外,石棉肺患者血清中IgM、IgG、抗核抗体和类风湿因子含量较高,肺内有异常球蛋白沉积,因而推测纤维化的形成,可能是巨噬细胞崩解,形成变性蛋白,引起自身免疫反应的结果。
【病理变化】
石棉肺的病变特点是肺间质弥漫性纤维化,其中可见石棉小体以及脏层胸膜肥厚和在壁层胸膜形成胸膜斑。因为吸入肺内的石棉纤维易随气流沿支气管长轴进入肺下叶,故肺病变以两肺下部为重,不同于矽肺病变以两肺中部为重的特点。
肉眼观,肺缩小、变硬,早期病变主要发生于两肺下部。由于细支气管周围、肺泡壁及小叶间隔内纤维组织增生,使肺呈明显的纤维网状结构。晚期,肺间质纤维化更广泛,更明显,肺组织陷于弥漫性纤维化。常因伴有明显的肺气肿和支气管扩张,肺组织可出现蜂窝状改变。肺胸膜尤其是下叶胸膜高度肥厚,晚期肺胸膜的纤维性增厚更广泛,甚至全肺均为灰白色的纤维瘢痕组织包裹。胸膜斑(pleural plaques)是指发生于壁层胸膜上的局限性纤维瘢痕斑块,境界清楚,凸出于胸壁,质地坚硬,呈灰白色、半透明而有光泽,状似软骨。胸膜斑常常位于两侧中、下胸壁,呈对称性分布。
镜下,早期病变为石棉粉尘引起的脱屑性间质性肺炎变化,表现为Ⅰ型肺泡上皮细胞受损,Ⅱ型肺泡上皮增生,后者呈立方形覆盖于肺泡表面。肺泡腔内有大量脱落的肺泡上皮细胞和巨噬细胞聚积。细支气管壁及其周围和血管周围的结缔组织中,以及肺泡间隔内有大量淋巴细胞和单核细胞浸润,有时也有一些嗜酸性粒细胞、浆细胞浸润。肺间质细胞增生,小动脉呈现闭塞性动脉内膜炎。随后,细支气管、血管周围及肺泡间隔内纤维结缔组织增生,到晚期则发展为肺组织弥漫性纤维化,大多数肺泡闭塞,由纤维组织填充。同时也可见一些衬以立方上皮的肺泡(腺样肺泡)。细支气管和小血管被包裹于纤维组织内,管壁纤维性增厚,管腔狭窄乃至闭塞。在增生的结缔组织纤维间散在着一些石棉小体(asbestos bodies),乃表面有铁蛋白沉积的石棉纤维,其长短不一(长者可超过100μm,短者仅数微米),黄褐色,呈哑铃状、分节状或蝌蚪形(图9-26),铁反应阳性。在石棉小体旁可见异物巨细胞。在弥漫性纤维化的肺组织中查见石棉小体是病理诊断石棉肺的重要依据。
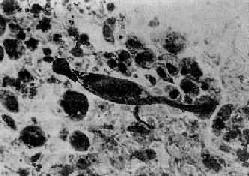
图9-26 石棉肺
图中央可见典型石棉小体,两端肥大,呈分节状,周围为尘细胞
【合并症】
动物实验和临床观察已证实,石棉属于促癌物质。近年来很多报道表明,石棉肺患者易并发肺癌、恶性胸膜和腹膜间皮瘤、食管癌、胃癌、结肠癌、喉癌等恶性肿瘤。据统计,石棉肺合并肺癌者可高达12%~17%,且多发于肺下叶。吸烟的石棉工人患肺癌的危险性比不吸烟人群高53~92倍。石棉肺合并恶性胸膜间皮瘤者也相当多见。有报告称,52例间皮瘤患者中约80%有接触石棉粉尘的职业史。从接触石棉粉尘到发现恶性间皮瘤的期限较长,平均42年。
石棉肺合并肺结核的频率约10%,远低于矽肺,而且多数病情较轻。此外,石棉肺易合并呼吸道感染、自发性肺气肿。晚期常并发肺源性心脏病和呼吸衰竭。
(三)农民肺
农民肺(farmer’s lung)是因吸入含有高温放线菌的有机粉尘而引起的一种外源性过敏性肺泡炎(extrinsic allergic alveolitis)。患者多系农业生产者,有经常接触发霉的稻草、麦秸和谷类等职业史。急性期时,典型病例在吸入含高温放线菌有机粉尘后4~8小时发病,出现畏寒、发热、呼吸困难或喘息、咳嗽、有少量粘液痰、乏力、全身肌肉酸痛、两肺可闻及湿啰音等症状和体征。急性期症状持续1~2天后可自行缓解,患者往往自认为是劳动后“受凉、感冒”,但也有的患者起病隐袭,呈现类似慢性支气管炎的症状。慢性期时,因反复发病,病变发展为弥慢性肺间质纤维化,并常并发肺心病,患者出现不同程度的心、肺功能障碍的症状和体征,重症患者可因呼吸衰竭和心力衰竭而死亡。农民肺的发病率,世界各地报告不同,一般为2.3%~8.6%。根据对湖北省洪湖县农民和城镇居民抽样调?
■[此处缺少一些内容]■
四、特发性肺间质纤维化
特发性肺间质纤维化(idiopathic pulmonary fibrosis,IPF)是指原因不明的弥漫性肺间质纤维化,为一种比较常见的肺疾病。可发生于任何年龄,患者以中、老年较多,多见于40~60岁之间。临床特征是出现进行性呼吸困难,X线显示两肺弥漫性网状结节状阴影,肺功能检查表明有限制性通气功能障碍、弥散功能障碍和肺的顺应性降低。病变特征是,早期表现为脱屑性间质性肺炎,晚期呈现不同程度的间质纤维化和蜂窝肺。多数患者呈慢性经过,但本病预后不佳,病死率甚高,常因肺功能不全和心力衰竭而死亡。平均生存期为5~6年,也有存活10年以上者。少数急性型病例进展急剧,多在6个月内死亡。年龄愈小者,病程愈短。
【病因和发病机制】
本病确切的病因和发病机制不明,可能与自身免疫、遗传因素和病毒感染有关。
目前较多的看法,认为特发性肺间质纤维化是一种自身免疫性疾病。其根据是本病常常与一些自身免疫性疾病,如类风湿性关节炎、硬皮病、系统性红斑狼疮等同时发生,而且在这些疾病中,肺部发生的病变与IPF的病变极其相似。IPF患者可出现高丙种球蛋白血症,升高的免疫球蛋白主要是IgG、IgM、IgA。部分病人可检出自身抗体,尤其是抗核抗体,类风湿因子阳性率较高。患者支气管肺泡灌洗液和血清中可检出免疫复合物(IgG-抗原复合物及IgM-C3b-抗原复合物),循环中免疫复合物也可在肺泡毛细血管壁内沉积。免疫复合物激活肺巨噬细胞释放趋化因子,引起肺组织内中性粒细胞、单核细胞和嗜酸性粒细胞浸润,而且这些细胞能产生氧自由基,还能分泌一些蛋白酶,如胶原酶、弹性硬蛋白酶等引起肺组织损伤,巨噬细胞还可产生纤维连接蛋白(fibronectin),促进纤维细胞增生,形成纤维化。
本病亦有家族性,有些患者是双胞胎,同家族的患者,蛋管异地居住多年,仍可发生同样的疾病。遗传连锁的研究表明,家族性IPF发生的危险与免疫球蛋白的γ异型有关。此外,在IPF患者中,存在HLA-B8、HLA-B12、HLA-B15和HLA-DW3、HLA-DW6、HLA-DR2抗原者增多,均提示本病发生与遗传因素有关。
约40%患者症状发作时有流感样表现及胸部症状,也有证据表明患者发病前有接触病毒史。有人报告,IPF患者血清中EB病毒抗体增加,可测出对病毒壳体抗原的IgA。提出EB病毒可能在IPF的病因学中起重要作用。
【病理变化】
本病早期,病变表现为脱屑性间质性肺炎,Ⅱ型肺泡上皮增生,肺泡腔内充满脱落的肺泡上皮细胞、肺泡巨噬细胞和淋巴细胞、中性粒细胞,有时可见透明膜形成。肺间质水肿,其中有灶性单核细胞、淋巴细胞和中性粒细胞浸润,以及广泛的纤维母细胞增生,肺泡间隔显著增宽。随着病情进展,肺泡腔和肺间质中的细胞逐渐减少。肺泡腔内炎性渗出物可发生机化,肺间质纤维组织逐渐增多,可发展至弥漫性间质纤维化。细支气管亦可见阻塞性细支气管炎病变,肺小动脉可呈现闭塞性动脉炎。细支气管和血管周围的纤维化也极为明显。到晚期,弥漫性纤维化使肺体积缩小、质地变硬,细支气管扩张呈小囊状,肺泡也不规则扩张成小气囊,小囊腔的囊壁破裂可形成较大的囊腔,这些囊被包裹于增生的纤维结缔组织之中,形成蜂窝肺。
【合并症】
特发性肺间质纤维化常合并肺部感染而导致呼吸衰竭。患者也常合并肺气肿、细支气管扩张和肺源性心脏病。部分患者可出现自发性气胸。少数病人可有肺性肥大性骨关节病。晚期特发性肺间质纤维化患者中,肺泡细胞癌、燕麦细胞癌、肺腺癌的发生率较高。
五、慢性肺源性心脏病
慢性肺源性心脏病(chronic cor pulmonale)是因慢性肺疾病引起肺循环阻力增加、肺动脉压力升高而招致的以右心室肥厚、扩张为特征的心脏病,简称肺心病。据流行病学调查,在我国肺心病的发病率较高,人群中的平均患病率为0.48%,尤以东北和华北地区较多,在各种器质性心脏病中,肺心病所占的百分比分别为18%~37%和12%~34%。
【病因和发病机制】
各种慢性肺疾病所致的肺循环阻力增加暨肺动脉高压是引起肺心病的关键环节。①慢性阻塞性肺疾病时,小气道的阻塞导致通气障碍,以及肺感染、肺间质纤维化及肺气肿均能破坏肺的血气屏障结构,减少气体交换面积,导致换气功能障碍。使肺泡气氧分压降低(缺氧),二氧化碳分压增高,引起肺小动脉痉挛(缺氧可干扰血管平滑肌细胞膜钾、钠离子交换和促使肥大细胞释放血管活性物质,引起肺小动脉痉挛)。②缺氧还能导致肺血管构型的改变,使肺小动脉中膜肥厚、无肌性细动脉的肌化,从而导致肺循环阻力增加和肺动脉高压。③限制性肺疾病,如胸廓病变、脊柱弯曲、胸膜纤维化及胸廓成形术后等,不仅可引起限制性通气障碍,还可压迫较大的肺血管和造成肺血管的扭曲,导致肺循环阻力增加暨肺动脉高压。④肺血管疾病,如反复的肺动脉栓塞和原发性肺血管疾病也可减少肺血管床面积而导致肺循环阻力增加和肺动脉高压。对增高的肺循环阻力,肺小动脉发生管壁平滑肌肌化增强,右心室也发生心肌细胞的适应性肥大,但右心室心肌细胞的适应能力是有限度的,当右心室负荷增高2~3.5倍时,极易出现心腔扩张。因此,肺心病可视为肺小动脉和右心室对慢性肺疾病引起的肺循环阻力和压力增高而发生的适应性反应,属于一种特殊的心脏病。
【病理变化】
1.肺部病变除原有的慢性支气管炎、肺气肿、肺间质纤维化等病变外,肺心病时肺内主要的病变是肺小动脉的变化,表现为肌型小动脉中膜肥厚、内膜下出现纵行肌束,无肌性细动脉肌化。还可发生肺小动脉炎,肺小动脉弹力纤维和胶原纤维增生以及肺小动脉血栓形成和机化。此外,肺泡壁毛细血管数量显著减少。
2.心脏病变右心室肥厚,心腔扩张,扩张的右心室使心脏的横径增大,并将左心室心尖区推向左后方,形成横位心,心尖主由右心室构成。心尖钝圆、肥厚。心脏重量增加(图9-27)。右心室前壁肺动脉圆锥显著膨隆(图9-28)。肥厚的右心室内乳头肌和肉柱显著增粗,室上嵴增粗,肺动脉圆锥处心肌壁增厚。通常以肺动脉瓣下2cm处右心室肌壁厚度超过5mm(正常约3~4mm)作为病理诊断肺心病的形态标准。镜下,可见心肌细胞肥大、增宽,核增大着色深。也可见缺氧所致的肌纤维萎缩、肌浆溶解、横纹消失,以及间质水肿和胶原纤维增生等现象。
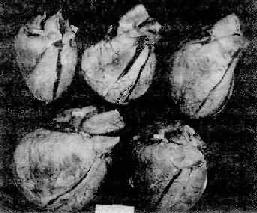
图9-27 正常与肺源性心脏病的心脏
图左上为正常成人心脏,其余为肺源性心脏病的心脏,其中最大者(左下)重785g
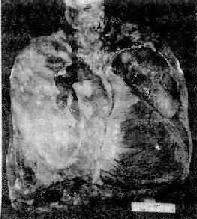
图9-28 慢性肺源性心脏病
两肺胸膜广泛性纤维性增厚,左肺上叶前段肺叶见肺大泡;左肺下叶被肥大转位的心脏压迫萎陷,右心室前壁肺动脉圆锥显著膨隆
【临床病理联系】
肺心病发展缓慢,临床表现除原有肺疾病的症状和体征外,主要是逐渐出现的呼吸功能不全和右心衰竭的症状和体征。受凉、上呼吸道感染、慢性支气管炎急性发作、肺炎及劳累等能诱发肺心病急性发作。每次急性发作都会进一步加重心、肺功能的损害,最后导致呼吸、循环衰竭。加强锻炼、增强体质,注意保暖并增强耐寒能力,戒烟,避开污染的空气,提高免疫力,预防诱发因素等对避免和减少肺心病的发生、发展至关重要。近年来,随着科学技术的进步,我国医务工作者正在从不同方面研究早期诊断肺心病的方法,如以X线检查、心电向量图和超声心动图检查,心、肺功能检查,放射性核素检查,肺阻抗血流图及肺微分阻抗血流图检查以及用漂浮导管技术直接测定肺动脉压力并进行血液动力学改变的研究和血液化学分析等,进行肺心病的早期诊断,将肺心病的防治工作提高到了新的水平。
六、肺不张和肺萎陷
肺不张(atelectasis)是指肺组织含气量过少,肺泡不能完全张开。肺萎陷(collapse of lungs)是指原已充满空气的肺组织因空气丧失而导致肺泡塌陷关闭的状态。
肺不张是先天性的,往往见于早产儿,由于呼吸中枢发育不成熟、分娩过程中引起的脑损伤或宫内窒息,使呼吸运动减弱或因细支气管被胎脂、胎粪、羊水阻塞,致出生时吸入的空气量不足而导致肺泡张开不全(新生儿肺不张)。肺发育不成熟,缺乏肺表面活性物质,使肺泡表面张力增加,肺的顺应性降低,也是导致新生儿肺不张的原因。肺萎陷是获得性的,多见于成人,有3种类型:①阻塞性(吸收性)肺萎陷:由于支气管被附近肿大的淋巴结或瘤块压迫,或支气管被肿瘤、异物、血凝块等阻塞,致空气不能进入所属末梢肺组织,其中原有的空气渐被吸收而导致一群小叶甚或整个肺叶无气和皱缩。②压缩性肺萎陷:大量胸腔积液,自发性气胸,严重的脊柱变形或腹腔巨大肿瘤或大量腹水使膈抬高,从而压迫肺组织,使肺泡陷闭而无气。③收缩性肺萎陷:由于肺组织广泛纤维化,纤维收缩所致。
七、呼吸窘迫综合征
(一)成人呼吸窘迫综合征
成人呼吸窘迫综合征(adult respiratory distress syndrome,ARDS)是由多种原因造成肺泡毛细血管膜弥漫性损伤和通透性增高而引起的一种急性低氧血症性呼吸衰竭。临床主要表现是患者突然发生进行性呼吸困难、紫绀、气促等急性呼吸窘迫症状,顽固性低氧血症和肺水肿,胸廓及肺的顺应性降低。X线检查显示迅速发展的两侧弥漫性肺浸润性阴影。成人呼吸窘迫综合征往往是严重创伤、大面积烧伤、大手术后、中毒、败血症或休克等患者严重的并发症。发病率有逐渐增高的趋势,病死率较高,据初步统计,其病死率为36%~68%。预后与肺病变的严重程度和肺功能状况有密切关系。
【病因和发病机制】
成人呼吸窘迫综合征是多种原因导致肺泡毛细血管膜(alveolocapillary membrane)急性损伤的结果。肺与外界相通,又是体内唯一接受全部心输出血量的器官,特别易受大气和血流中致病因子的伤害。而且,肺的血流量最多,毛细血管床的面积最大,受损的程度也比其他器官更为严重。致病原因如①肺感染;②吸入烟雾、毒气、胃内容物或溺水;③肺挫伤,放射性损伤;④氧中毒;⑤肺血栓栓塞,羊水栓塞,脂肪栓塞;⑥使用过量麻醉剂或某些药物(如水杨酸制剂、美散痛、海洛英、抗癌药等)以及⑦某些全身性病理过程(如败血症,脓毒败血症,休克,DIC,过敏反应,尿毒症,糖尿病酮症酸中毒,胰腺炎,严重创伤或烧伤等)和⑧某些治疗措施(如血液透析,体外循环,大量输出或输液)等都可引起肺泡毛细血管膜损伤,导致ARDS发生。其发病机制尚未完全阐明,可能主要与中性粒细胞和巨噬细胞在肺内聚积,释放大量蛋白水解酶(如胶原酶、弹性硬蛋白酶等)、氧自由基和一些血管活性物质(如前列腺素、白细胞三烯、血栓素A2等)引起肺泡毛细血管膜弥漫性损伤和通透性增高而导致肺水肿有关。此外,聚积在肺毛细血管内的血小板,既可阻塞毛细血管又可释放血管活性物质和溶酶体酶引起肺毛细血管膜损伤和通透性增高,在ARDS发病机制中起一定的作用。还有,肺表面活性物质减少,肺泡表面张力增加也是ARDS发病的因素之一。
【病理变化】
成人呼吸窘迫综合征的病理变化大致分为两个时期:
1.渗出期为病变早期,主要表现为肺间质毛细血管高度扩张、充血,肺间质和肺泡腔内有大量含蛋白质液体渗出(肺水肿)。在呼吸性细支气管及肺泡管或肺泡管与肺泡交界处的内表面有透明膜形成,其组成成分为蛋白质、纤维素和坏死的细胞碎屑。此外,肺间质中还可发生点状出血、灶性坏死以及淋巴管扩张。肺内往往发生局灶性肺萎陷。微血管内常有透明血栓和白细胞阻塞现象。
2.增殖期发病后数日,即转入损伤后的修复过程。Ⅱ型肺泡上皮细胞及肺间质的纤维母细胞大量增生,透明膜发生机化、纤维化,而且纤维化过程发展迅速。透明膜和肺泡间隔的纤维化很快扩展到全肺,导致弥漫性肺间质纤维化。
肉眼观,早期病变时肺表面湿润,有散在出血斑、出血点,并可见呈暗红色略凹陷的斑片状肺萎陷区。切面上,有大量泡沫状液体流出。3~4天后,肺出血、实变更加明显,呈牛肉样外观。可合并灶状梗死和支气管肺炎。
(二)新生儿呼吸窘迫综合征
新生儿呼吸窘迫综合征(neonatal respiratory distress syndrome,NRDS)指新生儿出生后已出现短暂(数分钟至数小时)的自然呼吸,继而发生呼吸过速、陷落呼吸(胸壁下陷)、呻吟、紫绀等急性呼吸窘迫症状和呼吸衰竭。患儿肺内形成透明膜为其主要病变特征,故又称肺透明膜病(hyaline membrane disease)。NRDS是新生儿发病率较高的疾病,主要见于妊娠不足35周龄的未成熟儿,糖尿病孕妇娩出的新生儿,体重过低的新生儿,剖腹产或臀位产的新生儿。此外,在第二个挛生儿也较常见。据报道,NRDS有家族性倾向,预后较差,病死率甚高。
【病因和发病机制】
新生儿呼吸窘迫综合征的病因与肺发育不全,缺乏肺表面活性物质有关。肺表面活性物质系由Ⅱ型肺泡上皮细胞合成和分泌,由磷脂酰胆碱、磷脂酰甘油等磷脂成分和蛋白质组成,具有降低肺泡表面张力,降低毛细血管内压和防止血浆滤出的作用。表面活性物质系统的发育至妊娠35周时才渐臻完善,其分泌达最高水平。肺发育不全,表面活性物质系统发育差(形态表现为Ⅱ型肺泡上皮细胞普遍缺乏板层小体),表面活性物质缺乏,使肺泡表面张力增加,肺顺应性降低,因而肺泡张开不全和不张。肺不张发生后,肺的通气和换气功能障碍导致缺氧、二氧化碳潴留和酸中毒,遂引起肺血管痉挛、肺血流灌注不足,从而损伤毛细血管内皮细胞,使毛细血管壁通透性增高,造成血浆蛋白漏出。同时,因缺乏表面活性物质,毛细血管内压增加,防止血浆滤出的作用降低,也促使血浆蛋白易于滤出。滤出到肺泡腔内的血浆蛋白凝集为透明膜,贴附于肺泡壁内表面,进一步影响气体交换,更加重了缺氧、二氧化碳潴留和酸中毒,肺血流灌注更为减少,进一步抑制肺表面活性物质的形成,如此形成恶性循环。
【病理变化】
肉眼观,肺含气少,色暗红,质韧,重量和体积可无明显改变。镜下,在呼吸性细支气管壁、肺泡管和肺泡壁上贴附一层均匀红染的膜状物(透明膜)。各肺叶均有不同程度的肺张开不全和不张,末梢气道中有水肿液。有些病例,肺间质和肺泡腔内出血较为明显。有的还可见吸入的羊水成分(鳞状上皮细胞和角化物质)。
八、肺癌
肺癌(lung cancer)是严重威胁生命的恶性肿瘤,为我国常见恶性肿瘤之一。半个世纪以来,世界许多国家和地区原发性肺癌的发病率和死亡率均有增加,尤以人口密度较高的工业城市更为突出。在欧美一些发达国家,肺癌的发病率和死亡率已升为各种癌症之首。在我国也有明显上升和继续上升的趋势。肺癌死亡率较高的地区主要在上海、天津、北京、辽宁、吉林、黑龙江等地。国内肺癌死亡率最高的城市是云南省个旧市,死亡率为70.62/10万。肺癌多发生于40岁以后,高峰发病年龄在70~79岁之间。男性患者多于女性,在我国男、女性比例为2.13:1。据西方国家统计,肺癌约占所有男性癌症的30%,而近年来,由于在西方国家女性吸烟者明显增多,女性肺癌患者的比例相应上升。
【病因】
肺癌的病因较为复杂,各国的研究表明,肺癌危险与下列因素有关:
1.吸烟国内外的大量研究证明,吸烟,尤其是嗜吸卷烟是引起肺癌的重要危险因素,吸烟者比不吸烟者的肺癌发生率高25倍,80%~90%的男性肺癌与吸烟有关。日吸烟量越大,开始吸烟的年龄越轻,患肺癌的危险越大。戒烟后患肺癌的危险性随戒烟时间的延长而逐渐降低。烟雾中含多种化学物质,如烟碱(居古丁)、多环芳烃碳氢化合物、镍、砷等与癌的发生有关,而细胞内的芳烃羟化酶(AHH)含量与吸烟者肺癌的发生呈正相关,肺癌患者芳烃羟化酶活性显著高于对照人群。苯并芘等多环芳烃碳氢化合物在芳烃羟化酶的作用下,转变为环氧化物,成为终致癌物,可与DNA共价结合,引起细胞的突变和恶性转化。
2.大气污染 在大城市和工业区肺癌的发病率和死亡率较高,与大气污染有密切关系。工业和生活中能源(煤、柴油、汽油等)大量燃烧后的烟尘及产生的工业废气和生活废气是造成大气污染的重要原因。污染的空气中含有苯并芘、二乙基亚硝胺和胂等致癌物。许多国家的调查表明,工业城市中肺癌的死亡率与空气中苯并芘的浓度呈正相关。
3.职业因素肺癌的发生与某些职业有关。橡胶工人、镍业工人、石棉工人、铀矿、锡矿、荧石矿的采矿工人以及接触含砷粉制剂者肺癌的发生率很高,这与长期接触某种化学致癌物和放射性物质有关。如云南锡矿中肺癌发病率特别高,井下矿工肺癌发生率达到435.44/10万,可能与矿井中氡、氡子体和砷等因素有关。
4.电离辐射大剂量电离辐射可引起肺癌已为许多事实证明。人群中电离辐射的来源为自然界和医疗照射。铀矿和锡矿工人肺癌的危险是α射线造成的。日本原子弹伤害幸存者中肺癌明显增多。而且辐射的中子和α射线引起肺癌的危险性比单独α射线要高。还有报告指出,接受放射线治疗的患者肺癌的发生率也明显增高。
肺癌的组织发生
绝大多数肺癌均起源于各级支气管粘膜上皮,源于支气管腺体或肺泡上皮细胞者较少。因而肺癌实为支气管源性癌(bronchogenic carcinoma),包括鳞癌、腺癌、小细胞癌和大细胞癌几种主要类型。肺鳞癌主要起源于段和亚段支气管粘膜上皮,后者在致癌因子作用下,经鳞状化生、异型增生和原位癌等阶段再演进为浸润癌。肺腺癌来自支气管的腺体。细支气管肺泡细胞癌究系源于细支气管分泌粘液的上皮或富含糖原的Clara细胞,还是来自Ⅱ型肺泡上皮细胞,尚未最后定论。小细胞肺癌来源于位于支气管粘液腺和支气管粘膜内的Kultschitzky细胞(嗜银细胞)属APUD瘤。近年来有不少的报道,认为所有类型的肺癌均来自呼吸道粘膜的干细胞,它可向多方向分化,因而也可出现混合型癌。
【病理变化】
肉眼类型肺癌的肉眼形态多种多样,根据其部位和形态可分为3种主要类型:中央型、周围型和弥漫型。从尸检例看,中央型多于周围型,约为3:1。但从肺癌手术切除标本看,周围型则多于中央型,这是由于受手术指征限制的缘故。
(1)中央型:癌块位于肺门部,右肺多于左肺,上叶比中、下叶多见。癌由段支气管以上至总支气管发生,浸润管壁使管壁增厚、管腔狭窄甚或闭塞;进一步发展时,癌瘤沿支气管纵深方向浸润扩展,除浸润管壁外还累及周围肺组织,并经淋巴道蔓延至支气管肺淋巴结,在肺门部融合成环绕癌变支气管的巨大癌块(图9-29),形状不规则或呈分叶状,与肺组织的界限不清,有时比较清晰。癌块周围可有卫星灶。有时癌块内也可见坏死空腔。
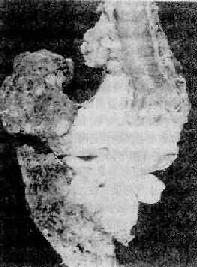
图9-29 右主支气管中央型肺癌
主支气管管壁增厚,埋没于巨大分叶状癌块中
(2)周围型:癌发生在段以下的支气管,往往在近脏层胸膜的肺组织内形成直径2~8cm呈球形或结节状无包膜的癌块,与周围肺组织的界限较清晰,而与支气管的关系不明显(图9-30)。本型发生肺门淋巴结转移较中央型为迟,但可侵犯胸膜。Pancoast瘤是位于肺上叶顶部的肺癌,可由胸膜长入胸壁。

图9-30 右上叶周围型肺癌
肿块位于肺叶周边部,呈结节状,其境界较清晰,但与支气管的关系不明显
(3)弥漫型:此型罕见,癌组织沿肺泡管、肺泡弥漫性浸润生长,很快侵犯部分大叶或全肺叶,呈肺炎样外观,或呈大小不等的结节散布于多个肺叶内(图9-31)。此时须与肺转移癌和肺炎加以鉴别。

图9-31 弥漫型肺癌
肺内满布无数灰白色小癌结节
近年来,国内外对早期肺癌和隐性肺癌问题进行了不少研究,因为这对于肺癌的早期发现和早期诊断具有重要意义。有人主张,早期肺癌可分为管内型、管壁浸润型和管壁周围型三型,但无淋巴结转移。日本肺癌学会将癌块直径<2cm并局限于肺内的管内型和管壁浸润型列为早期肺癌。痰细胞学检查癌细胞阳性,而临床及X线检查阴性,手术切除标本经病理检查证实为原位癌或早期浸润癌而无淋巴结转移者为隐性肺癌。
组织学类型 肺癌的组织结构多种多样,根据1976年世界卫生组织所定,肺癌的组织学类型可分为鳞状细胞癌、腺癌、小细胞癌、大细胞癌4种基本类型。据统计,仅40%~50%病例呈一致性组织构型,其余则在肿瘤的不同部位表现不同分化状态的组织构型,其转移癌的组织学类型也可与原发癌不同。
(1)鳞状细胞癌:为肺癌中最常见的类型,约占50%~70%,尸检统计占35%~45%。患者以老年男性占绝大多数,多有吸烟史。肿块生长较慢,转移较迟。因其多来自段以上或总支气管,故较易被纤支镜检查发现,痰脱落细胞学检查阳性率最高,达88.25%。组织学上分为高分化、低分化和未分化三型。高分化鳞癌癌巢中多有角化珠形成,低分化鳞癌癌巢中仅有少量或无角化现象(图9-32),未分化型癌细胞多弥漫排列,癌巢不明显,核分裂像较多。三型中以低分化型居多。
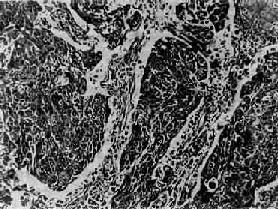
图9-32 肺低分化鳞状细胞癌
癌巢主由具有间桥的癌细胞构成,无角化珠
(2)小细胞癌:发生率在肺癌中居第二位(临床统计在40%以上,尸检统计占15%~25%)。患者男多于女(20:1),发病年龄约在35~60岁。小细胞肺癌亦多发生于肺中央部,生长迅速,转移较早,恶生度高,5年存活率仅1%~2%。小细胞癌的癌细胞很小,呈短梭形或淋巴细胞样,有些细胞呈梭形或多角型,胞浆甚少,形似裸核。癌细胞常密集成群,由结缔组织加工分隔(图9-33)。有时癌细胞围绕小血管排列成假菊形团或管状结构。小细胞肺癌起源于支气管粘膜和粘液腺内Kultschitzky细胞,是一种具有异源性内分泌功能的肿瘤。
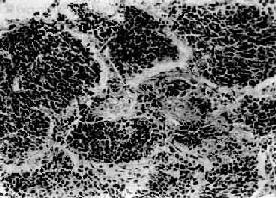
图9-33 小细胞肺癌
短梭形癌细胞平行排列,群集成团(燕麦细胞型)
(3)腺癌:发生率在肺癌中占第三位。临床统计占15%~20%,尸检统计占7%。患者女多于男(5:1),最常见于被动吸烟者。周围型肺癌中近60%为腺癌。肿块直径多在4cm以上,常累及胸膜,并常有肺门淋巴结转移。高分化腺癌癌巢呈腺管样结构(图9-34),可伴有粘液分泌;低分化腺癌的癌巢呈筛状或实体状;未分化腺癌的癌细胞呈高度异型性,可呈肉瘤样结构。
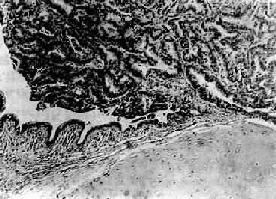
图9-34 肺高分化腺癌
腺癌自叶支气管粘膜发生,突入管腔内,癌组织主由腺样癌巢构成
肺腺癌的特殊类型还有腺样囊性癌、粘液癌、瘢痕癌和细支气管肺泡细胞癌等。瘢痕癌来自肺瘢痕组织,其中40%为腺癌,发展缓慢。细支气管肺泡细胞癌肉眼上呈弥漫型或多结节型。镜下,可见肺泡管和肺泡异常扩张,内壁衬以单层或多层柱状癌细胞,形成腺样结构,并常见乳头形成。而肺泡间隔大多保存完整。
(4)大细胞癌:癌细胞形成实体性癌巢或较大团块,主由胞浆丰富的大细胞组成,癌细胞高度异型。有时也可出现数量不等的多核癌巨细胞或胞浆空亮的透明细胞。此型恶性程度颇高,生长快,容易侵入血管形成广泛转移。
肺神经内分泌肿瘤(neuroendocrine tumors of lung)是指起源于APUD细胞,含有神经内分泌颗粒,能产生肽类激素的肺肿瘤,曾称为类癌。近20年来,随着电子显微镜和免疫组织化学技术的进展,虽然认识了肺神经内分泌肿瘤更多的形态和功能特征,但也带来了对该肿瘤诊断标准和分型的意见分歧,目前尚无统一意见。
1981年世界卫生组织将小细胞肺癌分为3个亚型:①燕麦细胞型:细胞呈短梭形或淋巴细胞样,②中间细胞型:细胞呈梭形或多角形,③混合型:小细胞癌伴有少量不典型鳞癌和分化不良的腺癌结构。1985年Palagudu将起源于Kultschitzky细胞的肺癌称为K细胞癌,并按其分化程度分为3个亚型:Ⅰ型:典型类癌,约有5.6%患者发生淋巴结转移,术后5年生存率高达95%;Ⅱ型:不典型类型,约有70%患者发生淋巴结转移或远处转移,术后5年生存率为65%;Ⅲ型:小细胞肺癌,是分化程度最低,恶性程度最高的一型,具有倍增时间(指肿瘤体积增大一倍所需的时间)短,浸润生长和广泛转移等特点,术后5年生存率<5%。各型肺神经内分泌肿瘤都具有表达神经肽类激素标志物的能力,表达的标志物可有蛙皮素、内皮素、胃泌素、促胃泌素释放肽、降钙素基因相关肽、血管活性肠肽等肽类激素和5-羟色胺、儿茶酚胺、乙酰胆碱等神经递质。此外,还含有神经元烯醇化酶和左旋多巴脱羧酶等物质。不同类型的肿瘤表达标志物的种类和数量虽有不同,但在同一种肿瘤中可有多种异源性肽类激素共存和肽类激素与神经递质共存的特点。鉴于肺神经内份泌肿瘤具有极其复杂的细胞表型和功能方面的异质性,而且应用电镜和免疫组化方法观察发现,肺神经内分泌肿瘤具有向APUD细胞和上皮细胞双向分化现象,特别是在小细胞肺癌中混杂的鳞癌和腺癌也可呈现上述各种标志物抗体免疫反应表达。提示不同组织学类型的肺癌可能有着共同的细胞起源。在同一肿瘤的细胞群体中出现形态和功能的异质性,可归因于体细胞异常转化的后代细胞所具有的多向性分化潜能。
【扩散途径】
1.直接蔓延 中央型肺癌常直接侵及纵隔、心包及周围血管,或沿支气管向同侧甚至对侧肺组织蔓延。周围型肺癌可直接侵犯胸膜,长入胸壁。
2.转移 肺癌发生转移较快、较多见。沿淋巴道转移时,首先至支气管肺淋巴结,再扩散至纵隔、锁骨上、腋窝、颈部淋巴结。血道转移常见于脑、肾上腺、骨以及肝、肾、胰、甲状腺和皮肤等处。
【临床病理联系】
肺癌的症状及其轻重与癌瘤的部位、大小和蔓延转移情况有关。肺癌一般发病隐匿,早期症状常不明显易被忽视。患者可有咳嗽、痰中带血、气急或胸痛。有时咯血,是最易引起注意而就医的症状。癌组织阻塞或压迫支气管时,可引起局限性肺萎陷或肺气肿。癌组织侵及胸膜可引起癌性胸腔积液;侵蚀食管可引起支气管食管瘘;侵犯纵隔内、气管旁淋巴结,压迫上腔静脉可引起上腔静脉综合征,表现为面颈部浮肿及颈、胸部静脉曲张。位于肺尖部的肺癌易侵犯交感神经链,引起病侧眼睑下垂,瞳孔缩小和胸壁皮肤无汗等交感神经麻痹综合征(Horner综合征);侵犯臂丛神经可出现上肢疼痛及手部肌肉萎缩。
肺癌,尤其是小细胞肺癌可有异位内分泌作用,可引起肺外症状。小细胞肺癌和腺癌可因5-羟色胺分泌过多而引起类癌综合征,表现为哮鸣样支气管痉挛,阵发性心动过速,水样腹泻,皮肤潮红等。肺性骨关节病(pulmonal osteoarthropathia)也是肺癌最常见的肺外症状,表现为伴有疼痛的骨、关节肥大和杵状指。此外,还可发生神经肌肉病变(肌无力综合征)、高血钙、低血糖和低钠血症、Cushing综合征及男性乳房发育症。这些肺外症状可在肿瘤切除后消失。所谓副肿瘤综合征(paraneoplastic syndrome)就是指除肿瘤及其转移灶直接引起的症状之外,伴随发生的由肿瘤引起的一系列异位激素性和代谢性症状综合征。肺癌、APUD瘤、胸腺瘤、肾透明细胞癌等都可引起副肿瘤综合征。
肺癌患者预后大多不良,早期发现、早期诊断和早期治疗至关重要。对于40岁以上的成人,特别有长期吸烟史并伴有咳嗽、痰中带血、气急、胸痛等症状者,或无痰干咳及与体位有关的刺激性呛咳的患者,必须提高警惕,及时进行X线、痰涂片细胞学(图9-35)和纤维支气管镜等检查,以及取活检组织作病理学检查,对肺癌的早期诊断具有重要价值。
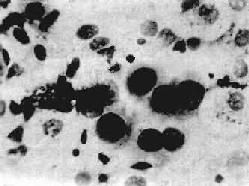
图9-35 痰涂片中的肺腺癌细胞
癌细胞圆形,核大、浓染,染色质粗,核仁大而明显,胞浆少